作者:陈美余,陶识丞,陈国生,唐弈遥,麦华明,广西医科大学口腔医学院/附属口腔医院
痣样
1.病例报告
患者男性,9岁,2019年7月因外院检查发现颌骨肿物2月收入院。患者家属述患儿因口内多颗牙不齐,替牙异常,2月前于外院就诊后行曲面断层片检查发现上下颌骨肿物,为行进一步治疗转诊至广西医科大学附属口腔医院就诊。
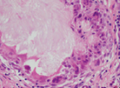
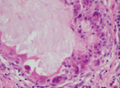
既往史:2016年7月21日在广西医科大学第一附属医院眼科全身麻醉下行“左眼眶内肿物摘除+结膜囊成形术”,术后病理提示眼眶及深部肿物为纤维结缔组织及脂肪组织,未见眼球组织,术前CT影像及术后病理结果见图1。

住院期间于2016年7月27日再次全身麻醉下行“左眼义眼座植入+眼窝再造术+异体巩膜移植术+睑裂缝合术”,术后诊断为“左眼先天性眼球缺如”“左眼先天性眼眶凹陷”,否认
专科检查:患者面容异于常人,头像照见图2,左眼缺失,眶距稍宽,额骨和颞顶骨较突出。双侧面部不对称,右面颊部鼻翼旁较左侧膨隆,面部及唇部无麻木等感觉异常。双侧颞下颌关节活动度一致,关节区无压痛,开口型开口度正常。面部皮肤多个棕黑色痣(图3),表面无毛且光滑,未突出于皮肤表面,直径约0.5~3mm。


口内检查(图4)示:黏膜无明显异常,舌活动无异常;混合牙列,萌出恒牙:16、15、14、11、21、21、26、31、32、36、41、42、43、46;在位乳牙:52、53、64、65、73、75;口内牙多颗龋坏,双侧颌下及颈部未及肿大淋巴结,无听力异常。右手拇指畸形,关节缺如,无法弯曲,双侧手掌均可见多个针刺样小凹陷(图5)。


辅助检查:锥形束CT(cone beam CT,CBCT)检查(图6)示上下颌骨多处囊性低密度影,界限清,考虑多发性颌骨囊肿,上颌累及右侧上颌窦,下颌累及体部,未波及升支,12、22缺失,13、33埋伏阻生;头颅


基因学检测:按标准采血流程,采集患者外周静脉血5mL,抗凝管储存,冰冻保存,送浙江安诺优达生物科技有限公司行外显子组高通量测序,结果显示患者的PTCH1基因23号外显子第3944个核苷酸(从DNA编码起始位点ATG算起)发生C>T杂合突变(c.C3944T),导致编码的第1315个氨基酸发生p.P1315L改变,由P(

治疗经过:1)口腔外科手术治疗——小囊肿手术单纯刮治术,大囊肿一期开窗减压术,二期刮治术,术中照见图9;术后病理(图10)确诊多发性牙源性角化囊肿(odontogenic keratocyst,OKC);2)术后1年复诊时发现上颌引流不畅,管道堵塞,管上段大量角化物,收治入院拟行二次手术,行颌骨囊肿刮治术,术后CBCT示与首次术前CBCT相比,左下后牙区及右下后牙区颌骨囊腔基本消失,下颌颏部囊腔新骨进一步形成,囊腔较前缩小,上颌骨囊腔边缘新骨形成效果较差,各时期CBCT影像见图11。



2.讨论
NBCCS的临床表现主要是多发性牙源性角化囊肿(odontogenic keratocyst,OKC)、皮肤基底细胞癌(basal cell carcinoma,BCC)及NBCCS相关的其他系统疾病表现,加强对该疾病的认识能提高NBCCS的诊断率及预后。OKC是NBCCS最常见的口腔表现,NBCCS患者中65%~100%发现了OKC,20岁以下的青少年中更为常见。
最常见于下颌骨,上颌骨OKC发生率虽较下颌骨低,但它们比下颌骨OKC更具有复发性和难治性。在本病例中,术后复查结果也反映了相似的问题,相同的手术方式,上颌囊肿术后囊腔边缘成骨效果差于下颌骨,上颌骨OKC更具难治性。OKC是这种疾病的常见标志之一,即使在没有并发其他临床表现的情况下,也应该引起接诊医生的高度怀疑,皮肤多发性BCC也是NBCCS最具特征性的表现之一。
青春期至35岁人群的发病率最高,40岁或40岁以上的白种人NBCCS患者中有90%的病例确诊BCC,黑种人NBCCS患者中这个比例占40%,而在韩国人NBCCS患者中占15.2%,这从侧面反映出BCC的出现与暴露在紫外线下有关。在本病例中未发现明显皮肤异常,但患者的面部皮肤可见多个散在的痣,需告知患者及家属注意避免日晒,做好防晒措施。此外,NBCCS还可能伴发其他系统相关疾病,需要多学科联合诊治。
2011年5月,第一届基底细胞痣综合征国际学术研讨会发布了一份关于诊断标准的共识声明,NBCCS诊断标准:1)1个主要标准和分子学确诊;2)2个主要标准;3)1个主要标准和2个次要标准。
主要标准:1)20岁以前出现BCC或BCC发生数量过多,皮肤类型与阳光照射量不相称;2)20岁以前出现OKC;3)手掌或足底凹陷;4)大脑镰板层钙化;5)髓母细胞瘤,典型的结缔组织增生;6)一级亲属确诊基底细胞痣综合征(basal cell nevus syndrome,BCNS)。
次要标准:1)肋骨异常;2)其他特殊的骨骼畸形(即脊椎畸形、脊柱后侧凸畸形、第四掌骨短、轴后型多指畸形);3)大头畸形;4)唇腭裂;5)卵巢/心脏纤维瘤;6)淋巴肠系膜囊肿;7)眼畸形(即斜视、眼距过宽、先天性白内障、
本病例临床诊断满足2个主要标准及2个次要标准。人类孟德尔遗传表型数据库(online Mendelian inheritance in man,OMIM)制定的各种遗传病、性状、基因的编号,简称MIM号。目前已明确的NBCCS(MIM号:109400)致病基因有3个,分别是位于9q22.32的PTCH1(MIM号:601309)、1q34.1的PTCH2(MIM号:603673)和10q24.32的SUFU(MIM号:607035)。
其中最常见的致病突变是PTCH1基因突变。对本病例的外显子测序发现,PTCH1、PTCH2、SUFU这3个基因均出现突变,但是仅PTCH1出现错义突变[23号外显子第3944个核苷酸发生C>T杂合突变(c.C3944T),导致编码的第1315个氨基酸发生p.P1315L改变],PTCH1、PTCH2和SUFU均存在同义突变。该患者的PTCH1错义突变位点已有文献报道(PMID号:15888139),文中描述该突变位点与非
该病例的错义突变位点c.C3944T可能是NBCCS的其中一个新的突变位点。值得注意的是,本文报道的病例伴先天性左眼缺失,文献中提到伴有PTCH1突变的NBCCS患者中,很少见到严重的眼部受累,如眼组织缺损、小眼畸形等。但是,PTCH1突变的先天眼缺陷NBCCS患者的眼部结构与有Patched突变的果蝇的眼部结构相似。
文献中已有证据表明,眼球发育缺陷与PTCH1突变相关。NBBCS发病人群更常见于白种人,不同人种发病特点有差异,对于其发病率也有不同的报道。对该疾病的认识不足,容易导致误诊或漏诊,且该综合征的多样表现形式也加大了诊断的困难,加深认识和了解至关重要。NBBCS的伴发症状和并发症给患者带来极大的痛苦,早期诊断能给疾病的有效预防和治疗带来更多机会。
来源:陈美余,陶识丞,陈国生,唐弈遥,麦华明.痣样基底细胞癌综合征伴先天性左眼缺失1例[J].华西口腔医学杂志,2022,40(02):240-245.
 纵隔大细胞神经内分泌癌1例CT影像
纵隔大细胞神经内分泌癌1例CT影像  张力性纵隔气肿影像表现及严重度分级
张力性纵隔气肿影像表现及严重度分级  迅速增大的肺部结节,首先考虑良性,确诊需要肺穿
迅速增大的肺部结节,首先考虑良性,确诊需要肺穿  肺隔离症:易误诊为肺癌的占位性病变,肺穿刺禁忌!
肺隔离症:易误诊为肺癌的占位性病变,肺穿刺禁忌!  肺段与肺内管道应用解剖
肺段与肺内管道应用解剖  肺转移瘤的十种不典型CT表现
肺转移瘤的十种不典型CT表现  肺内淋巴结的CT表现特点及与病理对照
肺内淋巴结的CT表现特点及与病理对照  肺实变与肺不张的CT鉴别诊断
肺实变与肺不张的CT鉴别诊断  医生现身说法,这五种忙帮不得!
医生现身说法,这五种忙帮不得!  北大教授:要真正让医务人员有阳光体面的收入!医
北大教授:要真正让医务人员有阳光体面的收入!医  为值夜班的医生护士鼓与呼:请给我们更多关注!
为值夜班的医生护士鼓与呼:请给我们更多关注!  广东拟取消医院用药数量限制,满足患者多样性需求
广东拟取消医院用药数量限制,满足患者多样性需求  博士、硕士入职就给精装房!又有医院不惜下血本招
博士、硕士入职就给精装房!又有医院不惜下血本招  历时7年之久,温医生宣判无罪!
历时7年之久,温医生宣判无罪!  重磅!四川发文:严禁限制医生多点执业
重磅!四川发文:严禁限制医生多点执业  与真人医生诊断一致性达96%:AI医生应用前景广阔
与真人医生诊断一致性达96%:AI医生应用前景广阔 



辽ICP备15019299号-3